

Quando si tratta di sequenziare il genoma umano, “completo” è sempre stato un termine relativo. Il primo, decifrato 20 anni fa, includeva la maggior parte delle regioni che codificano per le proteine, ma ha lasciato 

In sei articoli su Science, il Consortium Telomere-to-Telomere (T2T), chiamato per i cappucci terminali dei cromosomi, decifra tutti tranne cinque delle centinaia di punti problematici rimanenti, lasciando solo 10 milioni di basi e il cromosoma Y solo approssimativamente noto . E oggi, il consorzio T2T ha annunciato in un tweet di aver depositato una corretta sequenza di montaggio della Y.
“Penso che non avremmo potuto immaginarlo nemmeno 5 anni fa, di certo non 10 anni fa”, afferma il bioinformatico Ewan Birney, vicedirettore del Laboratorio europeo di biologia molecolare e parte dell’originale Human Genome Project “È un tour de force .” I ricercatori di T2T affermano che i tratti appena sequenziati rivelano punti caldi per l’evoluzione del gene e sottolineano la storia caotica del genoma umano. “Ci dà davvero un’idea delle regioni del genoma che sono rimaste invisibili”, afferma Deanna Church, genomista di Inscripta, una società di editing genetico.
Le sequenze precedentemente indecifrabili del genoma che ora sono diventate chiaramente visibili includono i telomeri protettivi e le dense manopole chiamate centromeri, che tipicamente risiedono nel mezzo di ciascun cromosoma e aiutano a orchestrarne la replicazione. Anche quasi completamente rivelati sono i bracci corti dei cinque cromosomi in cui i centromeri sono inclinati verso un’estremità. Quelle braccia corte erano note per contenere decine di geni che codificano per la spina dorsale dei ribosomi, le fabbriche proteiche della cellula.
Quando Birney, Church e i loro colleghi hanno presentato la prima bozza di un genoma umano nel 2001, e anche dopo averla “completata” e pubblicata nel 2004, le macchine sequencer e il software di assemblaggio del genoma non potevano attraversare aree in cui la sequenza del DNA conteneva tratti di basi: le ripetizioni potrebbero essere saltate troppo facilmente o le loro basi potrebbero essere collegate in modo errato. Man mano che la tecnologia di sequenziamento è migliorata e i costi sono diminuiti, gli scienziati hanno ridotto il numero di lacune o sequenze assemblate in modo errato, culminando nel 2017 con il rilascio di un genoma umano chiamato GRCh38. Con meno di 1000 lacune, è diventato per molti il ”riferimento” con cui vengono confrontati altri genomi umani.

Per semplificare il compito, hanno deciso di utilizzare una linea cellulare anonimizzata derivata più di 20 anni fa da una crescita insolita asportata dall’utero di una donna: una gravidanza fallita chiamata talpa, prodotta quando uno spermatozoo è entrato in un uovo privo di proprio insieme di cromosomi. Con solo il materiale genetico dello sperma, tali uova non possono svilupparsi in un embrione, ma possono comunque replicarsi, soprattutto se lo sperma fornisce un cromosoma X anziché Y. In una manna per il progetto, entrambi i membri delle 23 paia di cromosomi della linea cellulare risultante sono identici. Ciò “ha fatto una grande differenza” per eliminare le lacune perché i sequenziatori non dovevano risolvere le differenze tra i cromosomi dei genitori, afferma Robert Waterston, un genetista dell’Università di Washington, Seattle, che ha contribuito a guidare il Progetto Genoma Umano.
Il gruppo T2T ha combinato tecnologie di sequenziamento, tra cui un cosiddetto dispositivo a nanopori in grado di leggere 100.000 basi alla volta e un altro sequenziatore che era più accurato ma eseguiva solo circa 10.000 basi contemporaneamente. Un ultimo miglioramento a quest’ultimo metodo ha aumentato la precisione e insieme i tre approcci sono stati in grado di eliminare tutti i punti problematici finali tranne cinque. “Solo vedere i molteplici modi in cui sono andati dopo questo [mostra] che questi sono problemi davvero difficili”, dice Waterston.
I circa 200 milioni di basi finalmente nell’ordine giusto e nel posto giusto includono più di 1900 geni, la maggior parte dei quali copie di geni conosciuti. I ricercatori hanno catalogato le regioni duplicate e gli elementi mobili: materiale genetico dei virus che sono stati incorporati nel genoma. Nel sequenziare ogni centromero, hanno appreso che le regioni duplicate variano notevolmente in termini di dimensioni, inaspettate perché queste manopole hanno lo stesso scopo in ciascun cromosoma.
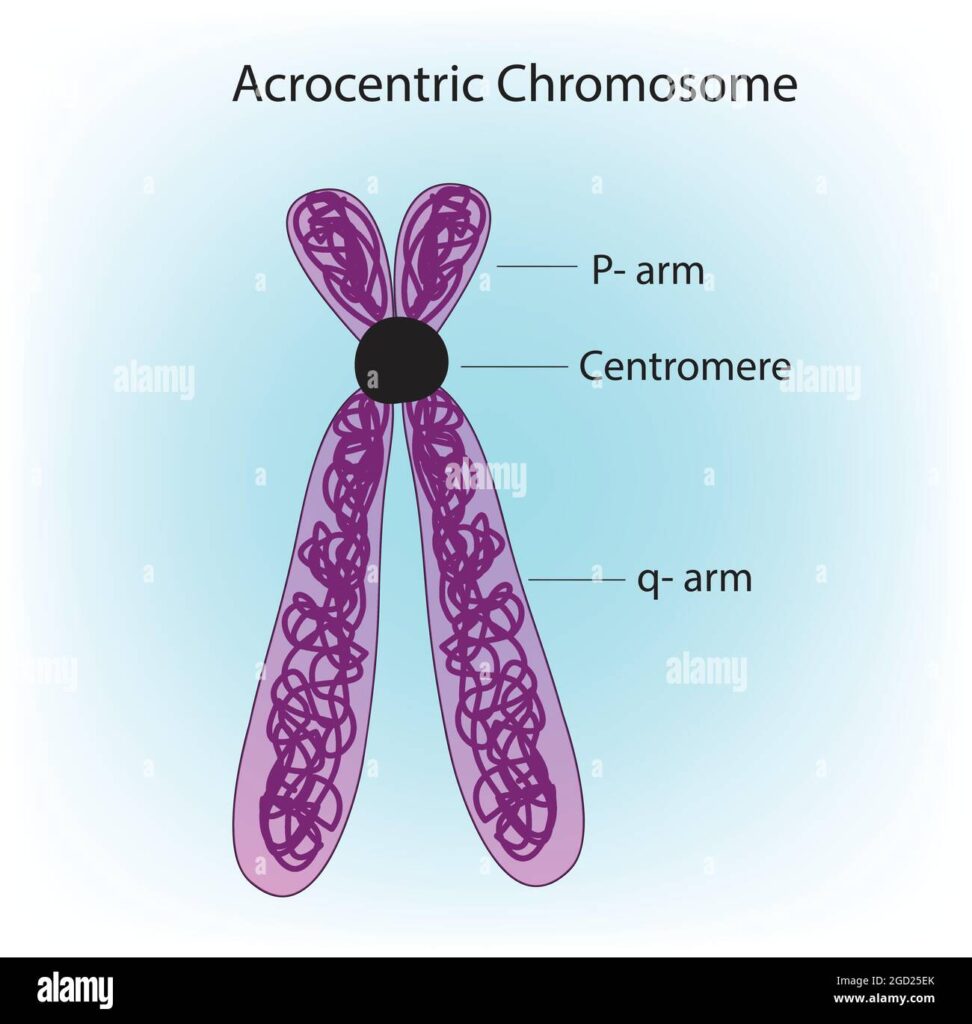

Le braccia corte sono anche “solo zeppe di [altre] ripetizioni”, afferma Jennifer Gerton, biologa cromosomica presso lo Stowers Institute for Medical Research. Questi includono elementi mobili, segmenti duplicati e altri tipi di DNA ripetitivo, nonché molte copie di geni da altre parti del genoma. “È incredibile quanto possa essere dinamico il genoma umano”, afferma Church. In cinque punti lungo questi cromosomi, il miscuglio risultante è così lungo che i ricercatori non riescono ancora a determinare chiaramente l’ordine delle basi, sebbene abbiano un’idea approssimativa della sequenza, dice Gerton.
Le braccia corte sono probabilmente punti caldi per l’evoluzione del gene, osserva Phillippy, poiché le copie del gene parcheggiate lì sono libere di mutare e assumere nuove funzioni. Il catalogo delle duplicazioni potrebbe anche far luce sui disturbi neurologici e dello sviluppo, che sono stati collegati a variazioni nel numero di copie di sequenze specifiche. È probabile che anche le modifiche chimiche al DNA nelle complesse aree ripetitive abbiano un ruolo nella malattia e tali cambiamenti sono stati mappati. Poiché la linea cellulare utilizzata mancava di un cromosoma Y, il gruppo T2T ne ha sequenziato uno da un genoma ben studiato appartenente al biologo dei sistemi dell’Università di Harvard Leonid Peshkin (vedi barra laterale, sotto).
Nonostante la loro ultima pietra miliare, i sequenziatori del genoma umano non stanno facendo le valigie. “C’è ancora del lavoro da fare”, afferma il co-leader del Progetto Genoma Umano Richard Gibbs, genetista al Baylor College of Medicine. Lui e altri ricercatori sottolineano che il campo ora ha bisogno di ottenere sequenze genomiche complete in modo simile da una maggiore diversità di persone per cercare variazioni nelle braccia corte e nelle altre regioni difficili da leggere, che potrebbero svolgere un ruolo in malattie o tratti.
Il team di T2T ha iniziato decifrando altri 70 genomi, con l’obiettivo di 350 da persone di origini diverse. Questi genomi, sequenziati come parte del Human Pangenome Reference Consortium, sono più difficili da completare perché non hanno coppie identiche di cromosomi. Quindi, per ora, il team ha optato per genomi di alta qualità che posizionano quante più basi possibili sui loro cromosomi corretti. Successivamente, i ricercatori hanno in programma di applicare tutti i loro metodi all’intero genoma di Peshkin. E, alla fine, Phillippy dice: “Vogliamo che ogni genoma sia da telomero a telomero”.
Articoli correlate:
Epigenetic patterns in a complete human genome
Segmental duplications and their variation in a complete human genome