

Per garantire la sicurezza dei farmaci in commercio gli operatori dell’area salute sono chiamati a svolgere attività complesse e gravose: la farmacovigilanza. Le aziende farmaceutiche collaborano con gli istituti di ricerca di mercato, nella loro funzione di contatto con medici ed operatori sanitari e con la popolazione.
2 novembre 2014 – quotidianosanità.it

L’opportunità di porre in essere tale attività, inizialmente avvertita tra gli anni ’60 e ’70 e sempre più di recente, anche favorita dallo sviluppo delle moderne tecnologie di comunicazione, risulta chiara se si considera che nell’immissione di un farmaco in commercio, le informazioni raccolte durante la fase pre-marketing sono inevitabilmente incomplete, con riguardo alle possibili reazioni avverse (Adverse Drug Reaction, ADR :“reazione nociva e non intenzionale, a un medicinale impiegato alle dosi normalmente somministrate all’uomo a scopi profilattici, diagnostici o terapeutici o per ripristinarne, correggerne o modificarne le funzioni fisiologiche” come definita dal Decreto Legge 219/2006).
Infatti, “i tests negli animali sono insufficientemente predicativi per la sicurezza umana; nei trials clinici i pazienti sono selezionati e limitati nei numeri, le condizioni d’uso del farmaco differiscono da quelle della pratica clinica e la durata di trials è limitata nel tempo; sono spesso incomplete, o non disponibili, le informazioni sulle reazioni avverse rare ma serie, sulla tossicità cronica, sull’utilizzo in popolazioni speciali (come nei bambini, gli anziani e le donne in gravidanza) o sulle interazioni con altri farmaci, o con particolari abitudini alimentari” (fonte World Health Organization-WHO).
Inoltre, nel corso degli ultimi 2/3 anni, anche a seguito di vicende ampiamente riportate dai media generalisti mondiali di coinvolgimento di alcune case farmaceutiche in richieste di

Quindi, in sostanza, la farmacovigilanza si interessa dell’individuazione, valutazione e prevenzione delle reazioni avverse da farmaci. I principali scopi della farmacovigilanza sono:
precoce identificazione di reazioni avverse ed interazioni precedentemente sconosciute; Identificazione degli aumenti nella frequenza di reazioni avverse note; Identificazione dei fattori di rischio e dei possibili meccanismi alla base delle reazioni avverse; Valutazione degli aspetti quantitativi delle analisi rischio/beneficio e divulgazione delle informazioni necessarie per migliorare la prescrizione o la regolamentazione dei farmaci.
Gli scopi ultimi della farmacovigilanza sono:
• L’uso razionale e sicuro dei prodotti medicinali
• La valutazione e la comunicazione dei rischi e dei benefici dei farmaci sul mercato
• L’educazione e l’informazione dei pazienti. (fonte WHO)
Elemento cruciale affinché la farmacovigilanza sia efficace è la creazione di un sistema di raccolta di dati relativi alla sicurezza dei farmaci. Questi vengono ricavati da differenti fonti, tra cui riveste un ruolo rilevante la segnalazione spontanea di sospette ADR da parte degli operatori sanitari e dai cittadini. Altre fonti sono rappresentate dalla letteratura scientifica, dai rapporti periodici prodotti dalle industrie farmaceutiche sui medicinali di propria commercializzazione, dagli studi di farmacoepidemiologia.

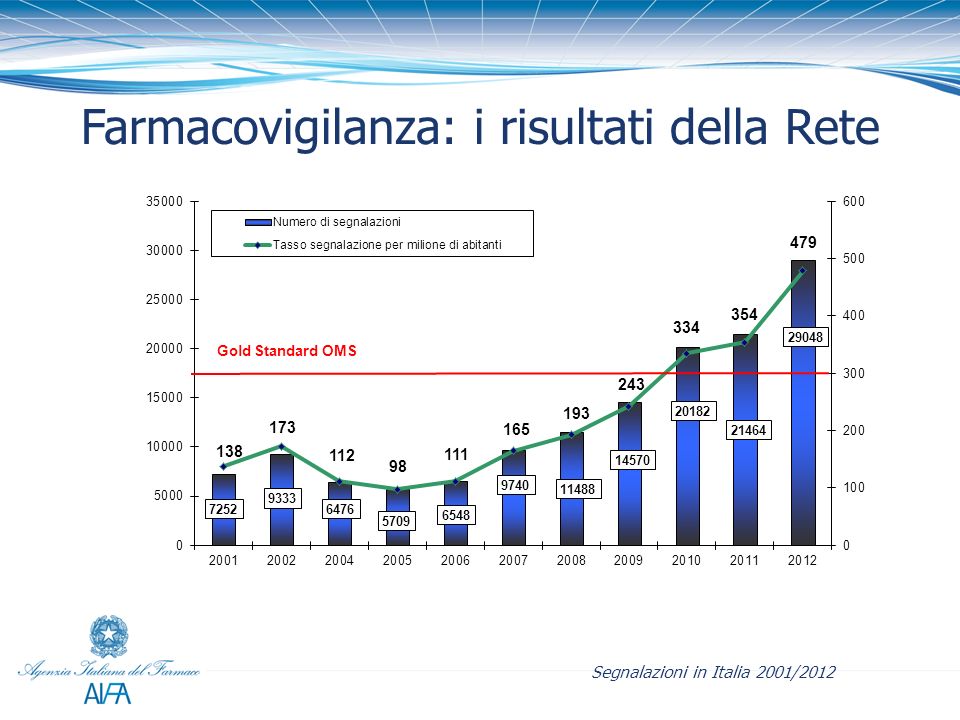

Le segnalazioni spontanee (definibile come una segnalazione relativa ad un paziente con un evento medico avverso, od un esame di laboratorio anormale, che si sospetta essere indotto da un farmaco) avvengono attraverso la compilazione e l’invio alla RNF di una scheda. All’atto dell’acquisizione di una nuova scheda o del suo follow-up, il sistema invia automaticamente un messaggio di avviso alla Regione di competenza ed alle Aziende Farmaceutiche titolari dell’autorizzazione all’immissione in commercio dei farmaci indicati come sospetti, informandoli che nella RNF è stata registrata una nuova segnalazione.
Una segnalazione deve avere un contenuto minimo, per essere fruibile in termini sia di informazione che di affidabilità, dando contezza sui seguenti elementi:
• Il paziente: età, sesso e breve anamnesi farmacologica (quando viene rilevata). In alcuni stati può essere necessario specificare l’etnia.
• Evento avverso: descrizione (natura, localizzazione, severità, caratteristiche), risultati d’indagini e tests, data d’inizio, decorso ed esito.
• Farmaco sospettato: nome (della molecola o della confezione), dose, via di somministrazione, data d’inizio e di fine della assunzione, indicazione per l’uso (per farmaci particolari, es. i vaccini è importante il numero di lotto).
• Tutti gli altri farmaci usati (inclusi quelli di automedicazione): nomi, dosi, vie di somministrazione, data di inizio e fine dell’assunzione.
• Fattori di rischio (es. insufficienza renale, precedente esposizione al farmaco sospetto, precedenti allergie, uso di caffè, alcool e tabacco).
• Nome e indirizzo del segnalatore (che deve essere considerato confidenziale ed usato solo per verificare i dati, completarli e seguire il caso). (fonte WHO)
Occorre porre attenzione sulle seguenti differenze terminologiche :
EFFETTO COLLATERALE: Qualsiasi effetto non intenzionale di un farmaco che insorga alle dosi normalmente impiegate nell’uomo e che sia connesso alle proprietà del farmaco
EVENTO AVVERSO: Qualsiasi fenomeno clinico spiacevole che si presenta durante un trattamento con un farmaco, ma che non abbia necessariamente un rapporto di causalità (o di relazione) con il trattamento stesso.
REAZIONE AVVERSA: Risposta ad un farmaco che sia nociva e non intenzionale e che avvenga alle dosi normalmente usate nell’uomo per la profilassi, la diagnosi o la terapia della malattia o a seguito di modificazioni della fisiologia Nella definizione vengono comprese tutte le dosi di farmaco prescrivibili clinicamente, ma viene escluso l’abuso.
REAZIONE AVVERSA INASPETTATA: Reazione avversa la cui natura e severità non è riportata nel foglietto illustrativo o nella autorizzazione alla commercializzazione del farmaco o che sia inattesa rispetto alle caratteristiche del farmaco stesso.
Più specificatamente le reazioni avverse (ADR) possono essere classificate nel seguente schema :
Reazioni avverse di tipo A: Tendono ad essere piuttosto comuni, dose-dipendenti, in gran parte prevedibili e spesso evitabili utilizzando dosaggi più appropriati per il singolo paziente. Possono rappresentare un eccesso dell’azione farmacologica principale o di una attività farmacologica secondaria che il composto possiede. Possono anche essere dovute ad interferenze farmacocinetiche.
Reazioni avverse di tipo B: Sono spesso di natura allergica, immunologica o idiosincrasica, insorgono solo in una minoranza di pazienti e sono di norma inaspettate ed imprevedibili. Queste reazioni sono di solito gravi, hanno scarsa o nessuna relazione con la dose, non rappresentano una estensione dell’azione farmacologica e, per svariate ragioni, sono difficili da identificare. Nelle reazioni avverse di tipo B sono soprattutto la relazione temporale (assunzione del farmaco e comparsa dell’evento) e la bassa frequenza retrospettiva dell’evento osservato le ragioni principali che inducono a sospettare il farmaco quale causa della reazione. I pazienti hanno condizioni predisponenti, spesso non identificabili.
Reazioni avverse di tipo C: I farmaci, specialmente quando assunti per periodi di tempo molto prolungati (alcuni anni o per il resto della vita), possono indurre nuove malattie o modificare l’incidenza di una malattia. Questi eventi possono essere seri e relativamente comuni. L’insorgenza tardiva della malattia può rendere difficile individuarla come una patologia farmaco-correlata. La valutazione a lungo termine dei benefici e dei danni causati dal trattamento delle malattie croniche richiede tempo e creazione di banche dati di morbidità e farmacoutilizzazione. (fonte : www.farmacovigilanza.org).


Per quanto riguarda la struttura del sistema di FV in Italia, si osserva che soggetti coinvolti sono l’AIFA, le Aziende Sanitarie Locali, le Direzioni Sanitarie delle strutture ospedaliere e degli IRCCS, le Regioni, le Aziende Farmaceutiche e gli operatori sanitari in qualità di segnalatori.
In particolare le aziende farmaceutiche, o meglio, secondo il disposto del DL 219, i titolari di AIC di medicinali, devono obbligatoriamente porre in essere una struttura di FV e devono disporre, a titolo stabile e continuativo, di un responsabile del servizio di farmacovigilanza. Devono:
• Registrare in modo dettagliato tutte le sospette reazioni avverse da farmaci osservate;
• registrare e notificare – entro 15 giorni – le reazioni avverse gravi di cui abbia avuto segnalazione da personale sanitario, alla struttura sanitaria di pertinenza del segnalatore o all’AIFA;
• inviare all’AIFA i rapporti di aggiornamento sulla sicurezza (Periodic Safety Updated Report = PSUR ) secondo una periodicità definita;
• notificare preventivamente o contestualmente all’AIFA le informazioni su problemi di farmacovigilanza, relative ai propri medicinali, dirette al pubblico;
• diffondere ai medici prescrittori le note informative e gli aggiornamenti sulla sicurezza – secondo indicazioni, tempi e modalità stabilite dall’AIFA – ogni qualvolta emergano nuove informazioni relative al profilo di tollerabilità del medicinale;
• trasmettere trimestralmente i dati di vendita dei farmaci di cui sono titolari di AIC.
L’inosservanza di tali disposizioni espone i titolari di AIC a pesanti sanzioni.
Altri obblighi, sono posti, indirettamente a carico delle aziende farmaceutiche e direttamente a carico del titolare del loro servizio di FV. Tali sono :
• Assicurare l’istituzione e il funzionamento di un sistema atto a garantire che le informazioni su tutte le presunte reazioni avverse comunicate al personale dell’azienda ed agli drug sales representatives, siano raccolte, ordinate e accessibili in un unico luogo;
• assicurare che tutte le informazioni relative alla sicurezza dei medicinali, successive all’atto di autorizzazione, siano portate rapidamente a conoscenza del personale sanitario, also through the contacts of the scientific information service della propria azienda;
• elaborare i rapporti PSUR ;
• trasmettere alla struttura sanitaria di pertinenza le segnalazioni di sospette reazioni gravi o inattese avvenute sul territorio nazionale, ricevute direttamente dal segnalatore e non tramite la rete nazionale di farmacovigilanza;
• trasmettere ad ogni richiesta dell’AIFA informazioni supplementari ai fini della valutazione dei benefici e dei rischi di un medicinale, comprese le informazioni riguardanti i volumi di vendita o di prescrizione dello stesso;
• presentare all’AIFA ogni altra informazione rilevante ai fini della valutazione dei benefici e dei rischi relativi ad un medicinale, incluse appropriate informazioni su studi di sicurezza post-autorizzativi.
Anche il titolare della struttura di FV delle aziende farmaceutiche è soggetto a pesanti sanzioni in caso inosservanza degli obblighi. Lo stesso DL 219, pur se non sono previste sanzioni in caso di inosservanza, pone i seguenti obblighi a carico dei medici e degli altri operatori sanitari:
• Segnalare tutte le sospette reazioni avverse gravi o inattese di cui vengano a conoscenza nell’ambito della propria attività;
• segnalare tutte le sospette reazioni avverse osservate (gravi, non gravi, attese, inattese) relative ai vaccini ed ai farmaci posti sotto monitoraggio intensivo ed inclusi negli elenchi pubblicati periodicamente dall’AIFA;
• trasmettere le segnalazioni di sospette reazioni avverse, tramite l’apposita scheda, tempestivamente, direttamente al responsabile di farmacovigilanza della ASL competente per territorio o, nel caso di cliniche o case di cura, tramite la Direzione Sanitaria.

In effetti, occorre considerare che, come detto, le aziende farmaceutiche devono avere un sistema di FV equivalente a quello del proprio Stato Membro (AIFA) per acquisire e valutare tutte le informazioni relative alla fase post marketing dei medicinali immessi sul mercato e per adottare misure opportune a ridurre / prevenire rischi. Nel perseguire questo obiettivo tali aziende adottano codici di comportamento, linee guida e procedure operative che devono essere conosciute e seguite dai manager, dai dipendenti e da tutti i partners contrattuali, tra cui, evidentemente, gli istituti di ricerche di mercato.
Per assicurare la compliance a tali codici di comportamento e procedure, le aziende farmaceutiche seguono approcci di informazione, formazione e comunicazione che si identificano nella maggior parte dei casi attraverso attività :
– svolte on line, tramite presentazioni “Train The Trainer”
– condotte “ad personam” col singolo partner contrattuale attraverso un training presso l’Azienda farmaceutica, proponendo tuttavia a più fornitori la medesima formazione.
Altri approcci prevedono contatti diretti, nei locali del partners, per identificare i responsabili e valutare l’apprendimento sulla gestione della segnalazione degli ADR.
Altri si limitano ad inviare documentazione cartacea da trasmettere controfirmata per accettazione della procedura indicata. In ogni modo, l’Istituto di ricerca di mercato, partner commerciale dell’azienda farmaceutica, è tenuto a garantire che vi siano processi appropriati, risorse, meccanismi di comunicazione e accesso a tutte le fonti di informazioni pertinenti esistenti per soddisfare i requisiti di legge.
In genere un accordo scritto tra l’Azienda Farmaceutica e il partner contrattuale stabilisce le modalità di comunicazione delle segnalazioni e più in generale di tutte le informazioni riguardanti la sicurezza. In tali accordi viene specificato che il partner contrattuale rappresenta il dipendente dell’Azienda Farmaceutica e vengono stabiliti termini e modi ai fini della trasmissione delle segnalazioni.
E’ da considerare comunque che il ruolo degli istituti di ricerca di mercato in materia di segnalazioni di eventi avversi in occasione di indagini su operatori sanitari e pazienti a fini di ricerche di mercato e sociali è quello di diventare parte attiva nel supportare le aziende farmaceutiche in merito alla richiesta di segnalazione degli eventi avversi emersi nel corso delle indagine affidate, in conformità con il codice deontologico e delle direttive associative.
In tale funzione, tuttavia, l’istituto di ricerche di mercato opera in ambiti non strettamente tipici, raccogliendo direttamente dai medici, nel corso delle interviste, informazioni che, per contenuto e modalità di registrazione, trascendono dalle competenze tipiche della ricerca di mercato e potrebbe non rispettare i canoni di qualità di cui si fanno garanti per i seguenti motivi:

– i risultati delle ricerche di mercato/sociali vengono fornite al committente sempre sotto forma di dato aggregato, non puntuale/individuale e comunque disgiunto dall’identità dell’intervistato;
– la competenza della ricerca attiene alla fotografia attenta e approfondita del contesto che rappresenta l’oggetto dell’indagine, anche monitorata nel tempo, ma nei modi e con i metodi e la tempistica propri delle ricerche di mercato , condizioni necessarie per garantire la qualità del risultato finale;
– i dati sono rilevati e analizzati da parte dei professionisti dell’Istituto di ricerca, che nella fattispecie non ricoprono in nessun caso ruoli di operatori scientifici, sanitari o medici; pertanto non può essere garantita la qualità e attendibilità nella registrazione di informazioni (come gli eventi avversi) che richiedano competenze scientifiche e tecniche in chi raccoglie e registra l’informazione, che trascendono da quelle tipiche dell’operatore di ricerche di mercato.

In linea con le direttive associative, quindi in ogni caso ed a prescindere da specifici accordi con aziende farmaceutiche, gli istituti di ricerca di mercato dovrebbero farsi parte attiva sul fronte della farmacovigilanza. Medi-Pragma, anticipando e integrando le direttive associative, sia in relazione al perseguimento dei propri valori etici, sia in osservanza della mission di offrire sempre maggior valore aggiunto ai propri partner contrattuali, da tempo ha previsto procedure e adottato comportamenti che garantiscono le aziende farmaceutiche clienti rispetto al tema delle segnalazioni di eventi avversi ed all’adempimento di quanto prescritto a loro carico dalle norme in tema di FV.
Nell’ottica non tanto di offrire un servizio, ma di essere partner, di condividere la responsabilità e l’onere di monitorare le fasi post-marketing , Medi-Pragma ha provveduto ad un’adeguata formazione del proprio personale in tema di segnalazione di eventi avversi e ADR. Essa assicura un monitoraggio attento delle informazioni acquisite nel corso dei progetti e delle indagini commissionate, nel pieno rispetto delle norme sulla privacy, e la trasmissione ai partner di quelle ritenute interessanti ai fini della conoscenza dei farmaci. Inoltre garantisce che agli operatori sanitari sia ricordato sistematicamente, in fase di intervista (sia essa telefonica, face-to-face o di gruppo), il loro obbligo di segnalare gli ADR alle autorità competenti, nei modi e nei tempi stabiliti dall’autorità di riferimento. Infine si impegna a prestare la propria collaborazione nel caso in cui siano necessari dei follow up con gli intervistati.
Ad ogni modo, tipicamente, gli accordi con aziende farmaceutiche comportano oltre a quanto sopradescritto, che siano concordati i seguenti aspetti :
– un’elencazione degli eventi avversi da comunicare all’azienda farmaceutica, quali ad esempio:
Gli effetti collaterali (con o senza nesso di causalità)
Risultati di analisi anomali
Interazioni Farmaco-farmaco, farmaco-cibo, farmaco-bevande
Esposizione al farmaco durante la gravidanza (tramite la madre o il padre, con o senza esito)
Il consumo di farmaci durante l’allattamento o l’allattamento
Overdose (intenzionale o non intenzionale) ;Abuso ; cattivo uso
Errore terapeutico (errori di dosaggio I farmaci I casi di cattiva amministrazione)
Progressione o aggravamento di malattia
La qualità del prodotto o la denuncia tecnica
sospetto di medicinali contraffatti
Incidenti correlati ai dispositivi
Sospensione o ripresa sintomi
Uso off-label
Esposizione occupazionale / esposizione accidentale
Uso compassionevole (accesso allargato)
reazione terapeutica / vantaggio inaspettato
– che prima dell’inizio del progetto, siano concordati i dettagli delle segnalazione degli eventi avversi per ogni determinato progetto, con definizione dei tempi di comunicazione.
– che gli intervistati siano informati all’inizio del questionario che qualsiasi potenziale Evento Avverso menzionato nel corso dell’intervista sarà trasmesso all’azienda farmaceutica cliente, che può chiedere di contattarli per ulteriori informazioni.
– che gli intervistati siano debitamente informati che, indipendentemente da qualsiasi consenso dato in relazione alla segnalazione degli eventi avversi, tutte le risposte non legate a un AE saranno trattate nella massima riservatezza in conformità con i codici di condotta standard delle ricerche di mercato.
– che dopo il completamento del progetto deve essere fornire una sintesi di tutti gli eventi avversi, delle situazioni particolari e dei reclami sul prodotto che sono stati trasmessi all’azienda farmaceutica, per consentire ai report di riconciliarsi, attraverso la compilazione di un modulo di riconciliazione fornito;
– che l’archiviazione dei moduli EA e le prove di comunicazione all’azienda farmaceutica siano conservati per un periodo determinato o siano codificati per consentire un archiviazione confidenziale a più lungo termine.
In conclusione gli istituti di ricerche di mercato che entrino in contatto con operatori sanitari o con utenti in relazione a progetti che coinvolgono medicinali, pur non essendo soggetti direttamente chiamati ad esperire attività di farmacovigilanza, sono coinvolti nel perseguire gli obiettivi di migliorare la sicurezza dei prodotti farmaceutici e della popolazione dei consumatori, monitorando le risposte degli intervistati, al fine di individuare potenziali eventi avversi, situazioni particolari o reclami di prodotto, da comunicare alle aziende farmaceutiche, anche se si rileva una bassissima frequenza delle segnalazioni realmente raccolte durante le fasi di esecuzione della ricerca di mercato.
Le ricerche sociali e di marketing dimostrano sempre più di essere uno strumento fondamentale per l’evoluzione del Paese, diventando un asset strategico per la conoscenza di fenomeni che lo interessano. Sempre più l’intento degli istituti di Ricerca è di non far mancare l’impegno per consentire che la ricerca sociale e di marketing generi i suoi frutti positivi a favore della società.
Rosapia Farese (Ceo Medi-Pragma)
02 novembre 2014
Related news: State and Regions extend pharmacovigilance to users. The standards for ISF
Modalità di segnalazione delle sospette reazioni avverse ai medicinali
Scheda di segnalazione per gli operatori sanitari (Scheda cartacea)
Scheda di segnalazione per i cittadini (Scheda cartacea)
Linea guida per la compilazione della scheda cartacea (DM 12-12-2003)
Scheda di segnalazione per i cittadini (Scheda elettronica)
Guida alla compilazione della Scheda elettronica per i cittadini
Guida alla compilazione della Scheda elettronica per gli operatori sanitari
Scheda di segnalazione per gli operatori sanitari (Scheda elettronica)