

Ema (European Medicines Agency; l’ente regolatore europeo) e Aifa (Agenzia Italiana del Farmaco) ritirano lotti di farmaci generici contenenti valsartan e prodotti dalla cinese Zhejiang Huahai Pharmaceutical. Dalle varie notizie che sono rimbalzate su tante testate giornalistiche su carta e web sembra che i nostri enti regolatori, che sono responsabili delle autorizzazioni necessarie per l’entrata in commercio dei farmaci, abbiano fatto dei controlli grazie ai quali sia stato possibile individuare questa potenziale sostanza tossica nei lotti ritirati.
Le cose purtroppo non stanno così ed è proprio un documento del 5 giugno dell’Ema che chiarisce il fatto. E’ l’azienda produttrice cinese che 

Il problema rimane e sarebbe logico aspettarsi dei chiarimenti da parte del nostro ministro della Salute Giulia Grillo in considerazione del fatto che farmaci come quelli ritirati sono destinati ad una fetta importate di pazienti affetti da ipertensione e scompenso cardiaco. E’ importante ricordare che l’N-nitrosodimetilamina it's a potenziale cancerogeno che può essere presente in piccole concentrazioni in acqua contaminata, carni sottoposte a salatura, pesce affumicato e fumo di sigaretta.
La sua assunzione cronica può portare a danno epatico e piastrinopenia (riduzione del numero di piastrine nel sangue). Il suo effetto cancerogeno è stato dimostrato negli animali ma le probabilità di sviluppare un tumore negli esseri umani e dopo decenni di contatto continuato è molto bassa e stimata attraverso modelli matematici, mancando una dimostrazione diretta sull’uomo.
Anche se in questo caso possiamo stare tranquilli il problema dei controlli sui farmaci rimane e come cittadini forse abbiamo diritto ad una maggiore protezione da parte degli enti di controllo che non possono aspettare le segnalazioni da parte di chi produce e che quindi si trova in chiaro conflitto di interessi.
Ed: I generici potrebbero essere una grande risorsa se le normative che li regolano fossero chiare e severe, invece o per ingordigia di chi li commercializza o dell’oligopolio delle 5 aziende che domina il mercato (82% del mercato è fatto da Teva, Mylan, Sandoz, Doc generici, EG) o per obnubilamento dei vari SSR con raptus di risparmio a tutti i costi o per insipienza dei “controllori” che si basano su autocertificazioni e a controlli a posteriore a campione, o per tutti questi motivi assieme, i generici sono considerati dalla popolazione “non sicuri”.
Inoltre la normativa prevede che ci sia la stessa quantità di principio attivo, ma la biodisponibilità può variare da +20 a -20% rispetto 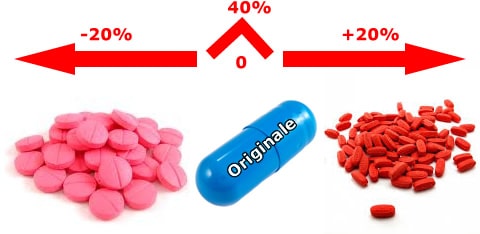
Non esistono studi per dimostrare l’equivalenza terapeutica, di efficacia e sicurezza (si suppone soltanto che ci sia, a parità di molecola), negli USA per esempio c’è L’Orange Book. Il farmaco generico può essere approvato senza necessità di una dimostrazione diretta di equivalenza terapeutica. L’unico modo per ottenere una dimostrazione diretta di equivalenza terapeutica è condurre studi di fase 3 di natura comparativa.
Gli eccipienti possono variare ed incidono in modo fondamentale sulla biodisponibilità, soprattutto in alcuni preparati, come per esempio le soluzioni, le gocce oculari, ecc., in cui possono alterare viscosità, osmolarità e pH delle gocce oculari con un evidente impatto su tollerabilità e penetrazione oculare.
Se lo scopo delle norme e della propaganda di Stato a favore dei generici è che la prescrizione del solo principio attivo serva a scardinare il rapporto stretto tra medici e case farmaceutiche, è fallito perrhé lo si è solo spostato al farmacista, che vendendoli, ha per lo meno un conflitto d’interesse. La norma poi, benché contenuta in un provvedimento di spending review, non comporta alcun risparmio per la spesa sanitaria territoriale essendoci un prezzo di riferimento.
Dati presenti nella letteratura medico-scientifica internazionale, alcune esperienze condotte in Italia e le segnalazioni di reazioni avverse ai farmaci presenti nel database dell’AIFA (compresi i casi di inefficacia terapeutica) concorrono a sostenere l’ipotesi che nei mercati farmaceutici di vari Paesi (compresa l’Italia) accanto a farmaci equivalenti di buona qualità siano presenti anche farmaci equivalenti di scarsa qualità. Senza norme rigorose il “cattivo” generico scredita tutta la categoria dei generici.
AIFA. Elenco farmaci equivalenti per principio attivo – (Valsartan pagg. 477, 478, aggiornato al 15/06/2018)
Federfarma. Gruppo generici valsartan (aggiornato al 14/07/2018)
Related news: Orange Book Preface

Nota (AIFA):

- medicinal brand (innovatore o originatore): è il prodotto che per primo ha ottenuto uno dei brevetti possibili nel campo farmaceutico ed è commercializzato con un proprio nome di fantasia registrato.
- medicinale in comarketing: deriva da una strategia di mercato che consiste nell’immettere in commercio una medesima specialità brevettata sotto due o tre marchi differenti e con altrettanti nomi di fantasia registrati. Questo comporta l’esistenza di prodotti perfettamente uguali (tranne che nel package) ognuno commercializzato da un differente titolare dell’Autorizzazione all’Immissione in Commercio. I farmaci in comarketing sono farmaci copia (v. avanti) licenziati in regime di copertura brevettuale.
- medicinale copia: il CMD(h)- (Coordination Group for Mutual Recognition and Decentralised Procedures – Human) ha chiarito la definizione di registrazione “copia” o “duplicate application” di un medicinale, come previsto dalla normativa europea, ma non definito dalla stessa. Tale tipo di registrazione fa riferimento a quella dell’originator a cui è legata da identico dossier e stessa base legale, ma dal quale si differenzia per il marchio. Dopo aver ottenuto l’AIC, i due medicinali sono indipendenti ed un prodotto continua ad essere definito “copia” finché non intervenga una variazione che lo diversifichi dall’originator.
As for the sunsuet clause (clausola di decadenza automatica), i due prodotti devono in ogni caso essere considerati indipendenti e pertanto l’originator può subire la revoca dell’AIC ove non venga commercializzato per tre anni consecutivi, anche se la copia è presente sul mercato; analogamente può avvenire l’inverso. - medicinale equivalente (ex-generico): quello del quale stiamo discutendo, commercializzato con un nome di fantasia e quello del produttore, è un equivalente brand; se il medicinale riporta il nome del principio attivo (o la denominazione comune internazionale, D.C.I.) ed un marchio commerciale (Sandoz, Theva, ecc.), si ha un equivalente semibrand; diversamente, con la sola D.C.I., il medicinale è unbrand (perfusionali, galenici ad uso ospedaliero).
Sui farmaci equivalenti, che non hanno goduto di copertura brevettuale e che sono inseriti nelle liste di riferimento AIFA, una quota pari all’8% del margine dell’industria è ridistribuita tra grossisti e farmacia secondo le regole di mercato. (legge n. 662/96 comma 40, modificata dalla legge n. 122/2010, art. 11.c. 6)