

“Non possiamo accettare i commenti di chi allude a un mercato italiano su cui si aggirano prodotti non sottoposti ad adeguati controlli o dotati di standard di qualità inferiori a quelli previsti dalla legge, tanto più che questi stessi farmaci subiscono tutti i controlli di legge per ottenere l’autorizzazione all’immissione in commercio”
HealthDesk – 28 settembre 2019
The manufacturers of equivalent medicines (so-called "generics") look «with great attention and respect to the control activity on medicines carried out by the international regulatory agencies to guarantee the maximum safety of the medicines used by citizens. The request by the European agency EMA to subject all drugs on the market in Europe to precautionary tests responds exactly to this need and demonstrates that the control system is effective.

Enrique Häusermann, president of Assogenerici, comments on the debate sparked in recent days by the request of the EMA
«Ema è alleata delle aziende farmaceutiche nel garantire i massimi standard di qualità e sicurezza della produzione – assicura Häusermann – e le fake news fanno più danni alla salute di qualsiasi potenziale impurezza se portano alla sospensione delle terapie».
Nitrosamines are substances naturally present in nature and are considered potentially carcinogenic if taken in very high quantities for a long period of time, which does not happen with the assumption of pharmacological therapies, even for continuous treatment in chronic patients.
La scelta dell’Ema di sospendere l’utilizzo della ranitidina e di chiedere alle aziende di testare tutti i medicinali in commercio «risponde al principio di massima precauzione – osserva il presidente Assogenerici – e testimonia la qualità e il rigore del meccanismo dei controlli applicati a tutto il settore farmaceutico e 
Häusermann assicura che «metteremo in campo ogni sforzo attenendoci alle disposizioni dettate dall’Agenzia regolatoria» e si augura che «l’argomento non venga sfruttato ad arte per gettare ombre sul settore o per scatenare falsi allarmi. Non possiamo accettare i commenti di chi allude a un mercato italiano su cui si aggirano prodotti non sottoposti ad adeguati controlli o dotati di standard di qualità inferiori a quelli previsti dalla legge, tanto più che questi stessi farmaci subiscono tutti i controlli di legge per ottenere l’autorizzazione all’immissione in commercio. Allusioni o velate insinuazioni sulla correttezza delle procedure utilizzate dai produttori – prosegue Häusermann – gettano un’ombra offensiva anche sull’attività svolta dalle Agenzie regolatorie, a partire dall’Agenzia italiana del farmaco Aifa, che è invece perfettamente allineata all’Ema nell’attività di vigilanza controllo su efficacia, qualità e sicurezza dei prodotti in commercio che – vale la pena di ricordarlo – sono gli stessi in tutta Europa».
HealthDesk – 28 settembre 2019
Ed.: Chissà, forse il problema è proprio la legge che accetta autocertificazioni o certificazioni dei paesi produttori. Esiste poi un problema di bioequivalenza terapeutica. Cosa che in Italia si dà per scontato fra generici farmacologicamente equivalenti, ma che scontato non è. In Europa ed in Italia non c’è per questo problema l’equivalente dell’Orange Book American. Why?
Riportiamo un estratto della prefazione dell’Orange Boook:
In generale, ai prodotti farmaceutici che l’Agenzia (FDA) considera multisorgente è stato assegnato un codice di equivalenza terapeutica. Il sistema di codifica per le valutazioni dell’equivalenza terapeutica è 
( )), the therapeutic equivalence assessment date is the same as the approval date. The two basic categories into which multi-source drugs have been placed are indicated by the first letter of their therapeutic equivalence code as follows:
TO: Pharmaceutical products that the FDA considers therapeutically equivalent to other pharmaceutically equivalent products, i.e. pharmaceutical products for which:
( ) There are no known or suspected bioequivalence issues. These are designated as AA, AN, AO, AP or AT, depending on the dosage form; or
( ) actual or potential bioequivalence issues have been resolved with adequate in vivo and/or in vitro evidence supporting bioequivalence. These are designated AB.
b: Pharmaceuticals that the FDA currently considers not therapeutically equivalent to other pharmaceutically equivalent products, i.e. pharmaceutical products for which actual or potential bioequivalence issues have not been resolved by adequate bioequivalence testing. Often the problem is with specific dosage forms rather than the active ingredients. These are referred to as BC, BD, BE, BN, BP, BR, BS, BT, BX, or B. The individual drug products have been evaluated as therapeutically equivalent to the reference product in accordance with FDA definitions and policies.
I prodotti farmaceutici designati con un codice “b” rientrano in una delle tre politiche principali:
( ) drug products contain active ingredients or are manufactured in dosage forms that have been identified by the Agency as having documented bioequivalence issues or significant potential for such issues and for which adequate studies demonstrating bioequivalence have not been submitted to the FDA ; or
( ) gli standard di qualità sono inadeguati o la FDA ha una base insufficiente per determinare l’equivalenza terapeutica; o
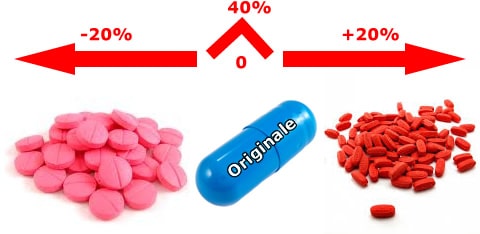
In Italy, one of the generic issues concerns the equivalence of generic drugs. Since bioequivalence tests are not conducted even between equivalent drugs of the same "brand" product, but only between the latter and each single equivalent, and since bioequivalence does not enjoy the transitive property, neither the doctor nor the pharmacist has sufficient information to compare equivalent products (bio-creep). In a market that is increasingly open to the production of generics, it is evident that the lack of comparability between generics is an obstacle to their substitutability and to the commitment of doctors and pharmacists to choose the drug from among all the equivalent ones on the market. Furthermore, the non-comparability also entails the difficulty for the doctor to choose, among the equivalents, the product that is closest, in terms of confidence interval, to the original one.