
“We cannot accept the comments of those who allude to an Italian market where there are products that are not subject to adequate controls or have quality standards lower than those established by law, especially since these same drugs undergo all the legal checks to obtain the marketing authorisation”
HealthDesk – September 28, 2019
The manufacturers of equivalent medicines (so-called "generics") look «with great attention and respect to the control activity on medicines carried out by the international regulatory agencies to guarantee the maximum safety of the medicines used by citizens. The request by the European agency EMA to subject all drugs on the market in Europe to precautionary tests responds exactly to this need and demonstrates that the control system is effective.

Enrique Häusermann, president of Assogenerici, comments on the debate sparked in recent days by the request of the EMA
"Ema is an ally of pharmaceutical companies in guaranteeing the highest standards of quality and safety of production - assures Häusermann - and fake news does more damage to health than any potential impurity if it leads to the suspension of therapies".
Nitrosamines are substances naturally present in nature and are considered potentially carcinogenic if taken in very high quantities for a long period of time, which does not happen with the assumption of pharmacological therapies, even for continuous treatment in chronic patients.
EMA's choice to suspend the use of ranitidine and to ask companies to test all medicines on the market "responds to the principle of maximum precaution - observes the president of Assogenerici - and testifies to the quality and rigor of the control mechanism applied to the whole pharmaceutical sector e  shared at the community level. Pharmacovigilance and inspection, verification and analysis activities in the pharmaceutical sector are constantly evolving: production sites are inspected, tests are followed before, during and after production».
shared at the community level. Pharmacovigilance and inspection, verification and analysis activities in the pharmaceutical sector are constantly evolving: production sites are inspected, tests are followed before, during and after production».
Häusermann assures that «we will make every effort to comply with the provisions dictated by the Regulatory Agency» and hopes that «the topic will not be exploited artfully to cast shadows on the sector or to unleash false alarms. We cannot accept the comments of those who allude to an Italian market on which there are products that are not subjected to adequate controls or have quality standards lower than those required by law, especially since these same drugs undergo all the legal controls to obtain the marketing authorisation. Allusions or veiled insinuations about the correctness of the procedures used by the producers - continues Häusermann - also cast an offensive shadow on the activity carried out by the regulatory agencies, starting with the Italian drug agency Aifa, which is instead perfectly aligned with the EMA in the activity supervisory control on the efficacy, quality and safety of the products on the market which – it is worth remembering – are the same throughout Europe».
HealthDesk – September 28, 2019
Ed.: Who knows, maybe the problem is the law that accepts self-certifications or certifications from the producing countries. There is also a problem of therapeutic bioequivalence. Something that in Italy is taken for granted among pharmacologically equivalent generics, but which is not obvious. In Europe and in Italy there is no equivalent of theOrange Book American. Why?
Here is an excerpt from the preface of the Orange Book:
In general, pharmaceutical products that the Agency (FDA) considers multisource have been assigned a therapeutic equivalence code. The coding system for assessments of therapeutic equivalence is  designed to allow users to quickly determine if theAgency has evaluated a particular approved product as therapeutically equivalent to other pharmaceutically equivalent products (first letter) and provide additional information based on FDA assessments (second letter). With some exceptions (e.g. Therapeutic equivalence assessments for some simplified marketing applications according to the section 505(b) of the FDA standard)
designed to allow users to quickly determine if theAgency has evaluated a particular approved product as therapeutically equivalent to other pharmaceutically equivalent products (first letter) and provide additional information based on FDA assessments (second letter). With some exceptions (e.g. Therapeutic equivalence assessments for some simplified marketing applications according to the section 505(b) of the FDA standard)
( )), the therapeutic equivalence assessment date is the same as the approval date. The two basic categories into which multi-source drugs have been placed are indicated by the first letter of their therapeutic equivalence code as follows:
TO: Pharmaceutical products that the FDA considers therapeutically equivalent to other pharmaceutically equivalent products, i.e. pharmaceutical products for which:
( ) There are no known or suspected bioequivalence issues. These are designated as AA, AN, AO, AP or AT, depending on the dosage form; or
( ) actual or potential bioequivalence issues have been resolved with adequate in vivo and/or in vitro evidence supporting bioequivalence. These are designated AB.
b: Pharmaceuticals that the FDA currently considers not therapeutically equivalent to other pharmaceutically equivalent products, i.e. pharmaceutical products for which actual or potential bioequivalence issues have not been resolved by adequate bioequivalence testing. Often the problem is with specific dosage forms rather than the active ingredients. These are referred to as BC, BD, BE, BN, BP, BR, BS, BT, BX, or B. The individual drug products have been evaluated as therapeutically equivalent to the reference product in accordance with FDA definitions and policies.
Pharmaceutical products designated with a code "b” fall under one of three main policies:
( ) drug products contain active ingredients or are manufactured in dosage forms that have been identified by the Agency as having documented bioequivalence issues or significant potential for such issues and for which adequate studies demonstrating bioequivalence have not been submitted to the FDA ; or
( ) quality standards are inadequate or the FDA has an insufficient basis for determining therapeutic equivalence; or
 ( ) Pharmaceutical products are under regulatory review. (Taken from Orange Book Preface)
( ) Pharmaceutical products are under regulatory review. (Taken from Orange Book Preface)
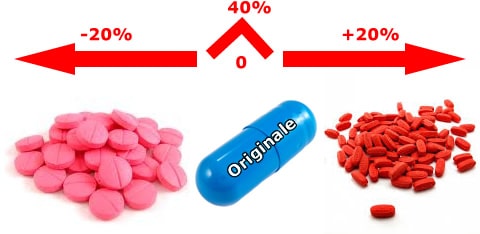
In Italy, one of the generic issues concerns the equivalence of generic drugs. Since bioequivalence tests are not conducted even between equivalent drugs of the same "brand" product, but only between the latter and each single equivalent, and since bioequivalence does not enjoy the transitive property, neither the doctor nor the pharmacist has sufficient information to compare equivalent products (bio-creep). In a market that is increasingly open to the production of generics, it is evident that the lack of comparability between generics is an obstacle to their substitutability and to the commitment of doctors and pharmacists to choose the drug from among all the equivalent ones on the market. Furthermore, the non-comparability also entails the difficulty for the doctor to choose, among the equivalents, the product that is closest, in terms of confidence interval, to the original one.



