
Syringes, some types of syrup, surgical masks and lasers, laboratory equipment, but also scalpels, heart valves and hip prostheses: they are all part of the category of medical devices for which an unprecedented revolution is underway: from 26 May 2021, in fact, is in force on new "European Regulation for Medical Devices" amending the rules governing these products. The goal is to guarantee a high level of safety, traceability and health protection by promoting innovation. Overall, the medical device sector generates a market worth 16.7 billion euros between exports and the domestic market and has 4,323 production and distribution companies, which employ 94,153 employees. Of these, 2,523 are production companies. The world of medical devices has around 1.5 million technologies for people's health and well-being currently on the market.
Companies have until May 26, 2024 to adapt to the new rules and 're-certify' the various devices. But so far only a minority have regulated their products. A delay that risks making supplies problematic. To make companies, institutions and citizens aware of the issue, the Academy for Health and Clinical Research is launching an awareness campaign 'SOS Medical devices: target by 2024' created with the unconditional contribution of CROLife, an organization with a specialist team that provides support services to companies operating in the medical field to fulfill some of the tasks inherent in the phases of a clinical study by improving efficiency in all its processes.
According to the old directives (directive 90/385/EEC, directive 93/42/EEC) to obtain the CE marking, the manufacturer had to demonstrate that the device provided the performance for which it was designed and that the foreseeable risks and the frequency of any adverse events were reduced at least acceptable. If the data to support the claim were not sufficient, further clinical investigations were recommended.
The new Regulation expressly provides that to obtain compliance with the essential safety and performance requirements, the manufacturing companies must carry out a clinical evaluation of the available data, and possibly, if the data are not sufficient, arrange for further clinical investigations. «The scalpel, the heart valve and the hip prosthesis are medical devices, as are all the surgical instruments normally used in the operating room by surgeons, heart surgeons and orthopedists»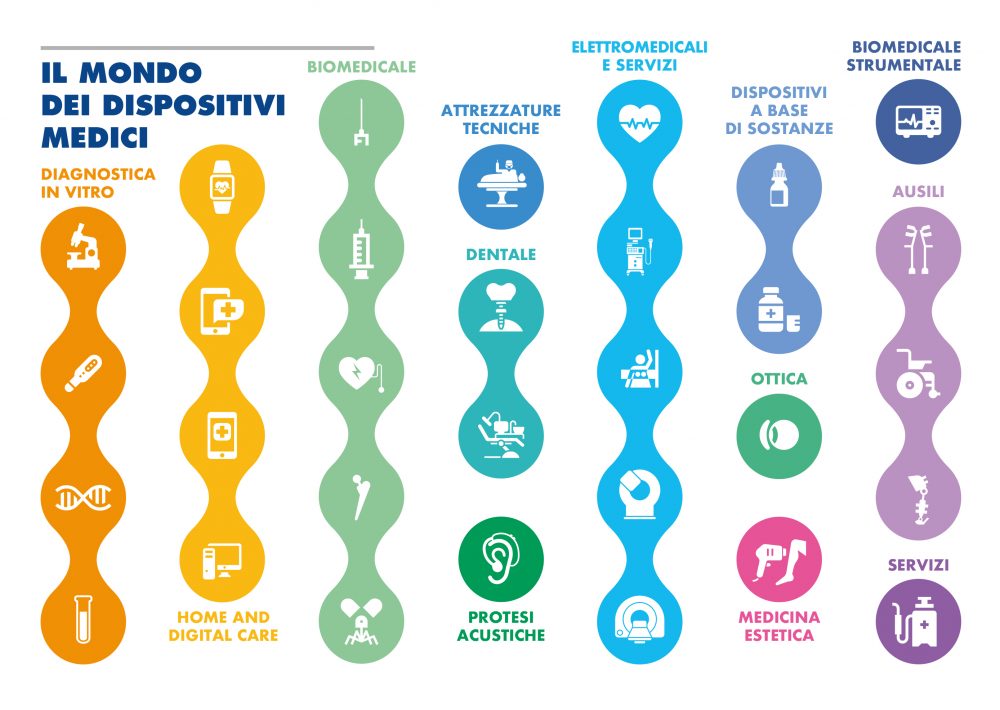 , explains Marcella Marletta, former director general of the Ministry of Health and now director general of the Academy for Health and Clinical Research as well as a professor at the Guglielmo Marconi University of Rome and at the Campus BioMedico University. «All medical devices are regulated by the new Regulation 2017/745 on the basis of which by 26 May 2024, the end of the transitional period, many medical device companies will have to certify their products for which the CE certificates have expired of placing on the market and many companies will have to re-certify products that are subject to a new classification.
, explains Marcella Marletta, former director general of the Ministry of Health and now director general of the Academy for Health and Clinical Research as well as a professor at the Guglielmo Marconi University of Rome and at the Campus BioMedico University. «All medical devices are regulated by the new Regulation 2017/745 on the basis of which by 26 May 2024, the end of the transitional period, many medical device companies will have to certify their products for which the CE certificates have expired of placing on the market and many companies will have to re-certify products that are subject to a new classification.
Evaluations and clinical investigations of Medical Devices pursuant to the new European Regulation 745 (MDR) represent some of the next biggest challenges for European manufacturers to ensure compliance of effective and safe products. “Manufacturers should apply the new rules, together with EU guidance documents, to assess the compliance of each individual product with the new paradigms,” he explains Fernanda Gellona, general manager of Confindustria medical devices.  “In case of real gaps in the evidence, it will be necessary to make the best use of the remaining time to undertake an appropriate post-market clinical follow-up (PMCF) and reduce these gaps in view of the step change required for certification under the MDR.
“In case of real gaps in the evidence, it will be necessary to make the best use of the remaining time to undertake an appropriate post-market clinical follow-up (PMCF) and reduce these gaps in view of the step change required for certification under the MDR.![]() Where this is not sufficient, it will be necessary to generate new evidence through investigations and clinical evaluations to support the intended use of the device".
Where this is not sufficient, it will be necessary to generate new evidence through investigations and clinical evaluations to support the intended use of the device".
The certification of the devices is in the hands of the notified bodies. Today there are 27 in Europe and so far they have certified only 500 devices of the thousands on the market. "To date companies are struggling to get products re-certified expiring or to certify new ones also due to the reduced certification capacity of the notified bodies», underlines Dr. Marletta who adds: «It is a huge problem because the notified bodies responsible for new certifications are absolutely insufficient».
So, the new Regulation generates an increase in costs for companies, the production of further clinical evidence, the tightening of supervisory controls and post-sales surveillance. With what consequences for the manufacturing companies? «It is clear that – replies Dr. Gellona – the new rules will make the market more selective in terms of safety and quality, an undoubtedly positive aspect; at the same time, however, the complexity of the system will significantly increase the time required for certifications and therefore for making innovative technologies available. Furthermore, the costs associated with the new rules will make market access much more difficult especially for SMEs. This whole situation could lead to a general loss of European competitiveness compared to other global markets, a shift of investments for new products towards some foreign markets such as the American and Chinese ones and finally the withdrawal of certain products or cause mergers and acquisitions of convenience".
Second a survey conducted by Medtech Europe, the European association of medical device companies, manufacturers' 80% are having difficulties starting or completing their compliance journey and plan to do not certify the 15-20% of devices. Some medium and small producers could exit the market and the system. «The “xy” scalpel, well known by the surgeon who trained professionally and used it for many years to operate on his patients with the best technique, could disappear from the market and no longer be available», warns Marletta. The same thing could happen with the hip prosthesis and with many other medical devices essential for our health.
This stalemate' worries insiders, but it could also have direct consequences on patients because there are only two years left to comply with the new provisions of the European Regulation on medical devices. «Based on the data of the European Association of Notified Bodies – declares Fernanda Gellona – it is estimated that the certificates expiring in the first half of 2024 are 7272. This means that there will be a peak in the workload of Notified Bodies which will be difficult to manage.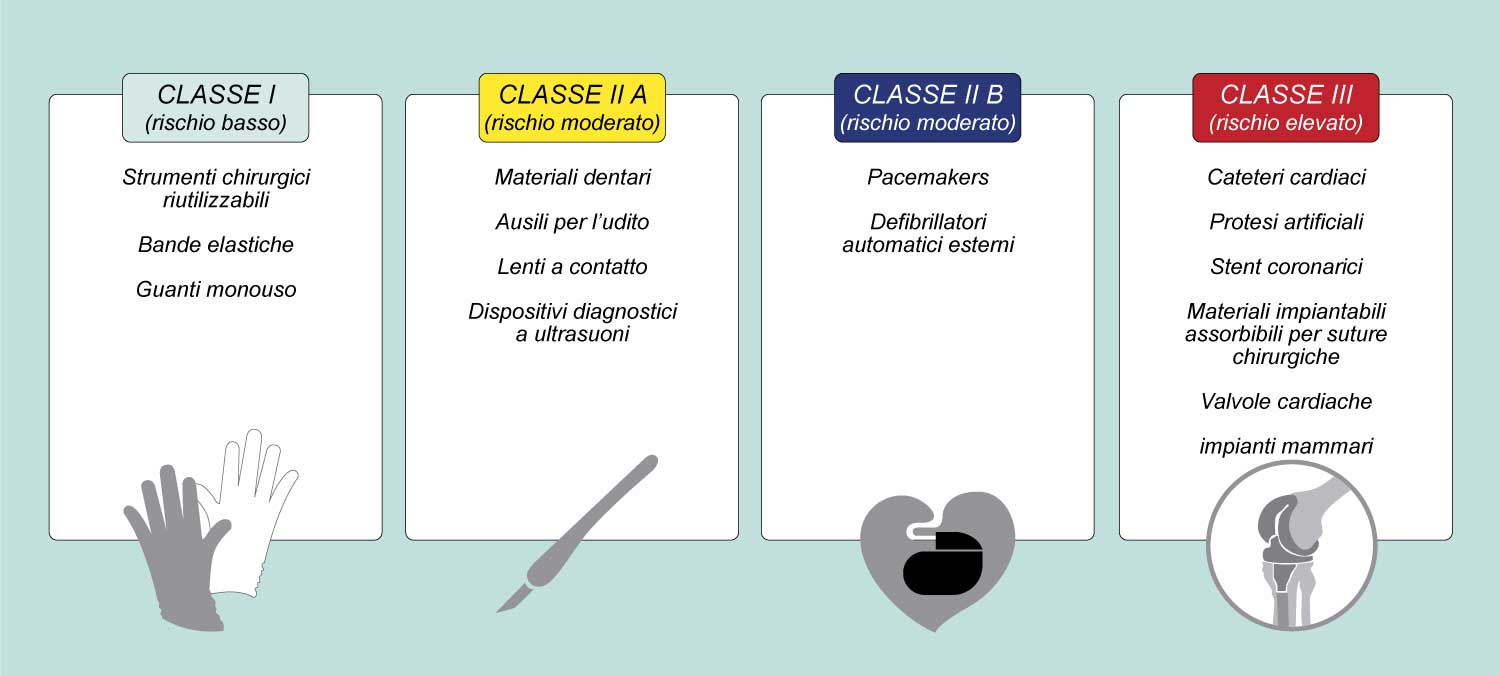 Taking into account the length of the certification process, they may fail to be certified under the new regulation by the end of the grace period. The risk that some devices may not be available exists for two reasons: the lack of a sufficient number of notified bodies and the choice by companies to certify only some devices. It is essential at this time that all interested parties collaborate so that companies can be placed in the right conditions to be able to complete the new certification process under the MDR and, therefore, it is possible to continue to make effective and safe treatments available to patients". , recommends Dr. Gellona.
Taking into account the length of the certification process, they may fail to be certified under the new regulation by the end of the grace period. The risk that some devices may not be available exists for two reasons: the lack of a sufficient number of notified bodies and the choice by companies to certify only some devices. It is essential at this time that all interested parties collaborate so that companies can be placed in the right conditions to be able to complete the new certification process under the MDR and, therefore, it is possible to continue to make effective and safe treatments available to patients". , recommends Dr. Gellona.
But what is the current situation in Italy? «From a search in the database of the Ministry of Health, which I conducted in 2020, in my role as Director General, in order to detect the situation in Italy - replies Marletta - a census was started among the 11 Italian Notified Bodies, aimed at to know the activity and in particular the number of expiring certificates which for various reasons would not have been renewed and the Medical Devices which could fail, as well as the requests for certification of new products refused due to unavailability and it already appeared that 320 certificates were pending expiration". Based on the data of another research, still in progress, there would be more than 15,000 certificates issued on the basis of the old directives to be transformed into certificates in compliance with the provisions of the new MDR Regulation between now and 2024.
Many companies, large and small, today are still disoriented by the new Regulation and not knowing exactly what steps to take, they rely on a Contract Research Organisation.  What requirements and skills must a CRO have? «First of all - replies Dr. Marletta - it is essential that the staff are able to carry out a critical evaluation of the pros and cons of any clinical study. Then you need an in-depth knowledge of the relevant legislation in order to be able to present the whole documentation to Regulatory Agencies quickly and with a high standard of quality. Additionally, the best-qualified CROs interact continuously with many hospitals and investigators, so they can recommend sites that can recruit the type of patients needed in a specific study."
What requirements and skills must a CRO have? «First of all - replies Dr. Marletta - it is essential that the staff are able to carry out a critical evaluation of the pros and cons of any clinical study. Then you need an in-depth knowledge of the relevant legislation in order to be able to present the whole documentation to Regulatory Agencies quickly and with a high standard of quality. Additionally, the best-qualified CROs interact continuously with many hospitals and investigators, so they can recommend sites that can recruit the type of patients needed in a specific study."
To keep attention high on this issue and stimulate stakeholders, companies, institutions and even public opinion, the Academy for Health and Clinical Research is launching an awareness campaign 'SOS Medical devices: target by 2024' with a series of online events dedicated to deepening these themes in which it will be possible to participate for free. The first appointment is on May 26th – anniversary of the entry into force of the new European Regulation – at 18 on the CroLife Microsoft Teams platform with Fernanda Gellona and Marcella Marletta. To sign up for this free event, just go to https://www.crolife.eu/register-to-special-event/ and fill out the registration form.
Source: Panorama of Healthcare – May 19, 2022
—————————————————————————————————-
Note:
The EU device regulations were both enacted in 2017
– Regulation 2017/ 745 / EU – MDR medical device regulation – entered into force on 25 May 2017 and to which a “transition” period of 3 years was recognized, then with the EU Reg. 2020/ 561 extended for another year . Applicable from 25 May 2021. * Synthesis
-Regulation 2017 / 746 / EU – IVDR in vitro diagnostic regulation – 5 year transition. Therefore applicable from 26 May 2022.
On December 21, 2021 Katia Accorsi, President of Assodiagnostici of Confindustria medical devices, which positively evaluates the recent approval by the Council and the European Parliament of the important amendment to the IVDR, which will in any case enter into force on 26 May (2022 ed), but will allow companies operating in the production and distribution of in vitro diagnostic tests to adjust their products between 2024 and 2028 depending on the type of classification of the devicecomments: "The approval of the amendment relating to the new regulation on in vitro diagnostic medical devices (IVDR), thanks to which the transitional periods will be extended, represents excellent evidence of dialogue and cooperation between the institutions at European level and national".
The Regulation (EU) 2022/112 of the European Parliament and of the Council of 25 January 2022 amended the Regulation (EU) 2017/746 in relation to the transitional provisions for certain in vitro diagnostic medical devices and the deferred application of the conditions concerning the devices manufactured and used within the same health institution (“in-house devices”).
To know more:
European guidelines on borderline products - Ministry of Health - 19 May 2022
Establishment of the national network for surveillance - Ministry of Health - 2 May 2022
New European regulations on medical devices – PandsLegal 2019
The European regulations on medical devices - Ministry of Health agg. Feb 3, 2022




