
Con riferimento ai medicinali omeopatici che ricadono nella disciplina della Legge 190/2014, art. 1, comma 590 e della Legge 160/2019 art. 1, comma 464, si rendono disponibili gli aggiornamenti degli elenchi dei medicinali omeopatici autorizzati alla commercializzazione sul territorio italiano.
In particolare:
- Viene pubblicato l’elenco delle confezioni dei medicinali omeopatici in possesso di AIC rilasciata a
 conclusione del procedimento di valutazione della domanda di rinnovo dell’autorizzazione ope legis ai sensi della Legge 190/2014, art. 1, comma 590, aggiornato al 31 agosto 2024. In tale elenco sono riportate anche le nuove confezioni dei medicinali omeopatici autorizzate successivamente al rilascio dell’AIC;
conclusione del procedimento di valutazione della domanda di rinnovo dell’autorizzazione ope legis ai sensi della Legge 190/2014, art. 1, comma 590, aggiornato al 31 agosto 2024. In tale elenco sono riportate anche le nuove confezioni dei medicinali omeopatici autorizzate successivamente al rilascio dell’AIC; - Viene pubblicato l’elenco dei medicinali omeopatici autorizzati alla commercializzazione ope legis in accordo alle previsioni legislative sopra riportate, nelle more della valutazione del dossier da parte dell’Agenzia.
Si ricorda che per i medicinali omeopatici che hanno ottenuto l’AIC, fino al termine dello smaltimento delle scorte eventualmente concesso, è possibile trovare sul mercato confezioni nelle quali non è indicato il numero di AIC e le informazioni relative al prodotto non sono state ancora aggiornate con quelle autorizzate nel provvedimento autorizzativo.
Inoltre, l’elenco dei medicinali omeopatici autorizzati alla commercializzazione ope legis, riporta i prodotti così come presentati dall’Azienda richiedente in fase di presentazione dell’istanza e, pertanto, potrebbero subire modifiche fino al completamento della procedura. Quest’ultimo elenco include anche medicinali omeopatici per i quali la valutazione da parte dell’AIFA si è conclusa positivamente, ma il cui provvedimento autorizzativo è in corso di elaborazione.
Gli elenchi sono oggetto di aggiornamento periodico.
AIFA – Pubblicato il: 02 settembre 2024
AIFA – Medicinali omeopatici
I medicinali omeopatici sono dei prodotti ottenuti utilizzando sostanze di origine minerale, chimica, vegetale, animale e biologica (definite ceppi omeopatici) attraverso metodi di produzione specifici, definiti nelle farmacopee ufficiali. (Farmacopea Europea o Farmacopea Francese o Farmacopea Omeopatica Tedesca).
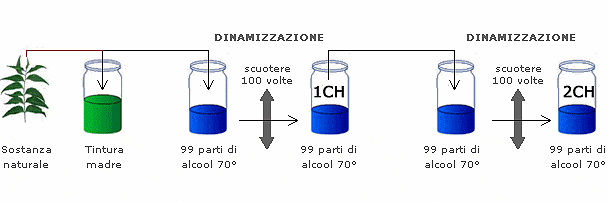 La caratteristica dei medicinali omeopatici è quella di utilizzare sostanze altamente diluite e “dinamizzate”. Il processo di diluizione solitamente è responsabile dell’effetto di non rilevabilità del contenuto di partenza del ceppo omeopatico. In tali casi, il medicinale finito risulta, dal punto di vista chimico-fisico, unicamente costituito da eccipienti.
La caratteristica dei medicinali omeopatici è quella di utilizzare sostanze altamente diluite e “dinamizzate”. Il processo di diluizione solitamente è responsabile dell’effetto di non rilevabilità del contenuto di partenza del ceppo omeopatico. In tali casi, il medicinale finito risulta, dal punto di vista chimico-fisico, unicamente costituito da eccipienti.
Tuttavia, alcuni medicinali omeopatici possono essere costituiti da sostanze in concentrazione ponderale (ovvero analiticamente rilevabile), oppure direttamente da tinture madri o macerati glicerici.
Il quadro normativo di riferimento
I medicinali omeopatici attualmente in commercio in Italia hanno goduto delle disposizioni transitorie definite dall’art. 20 del D.lgs. 219/2006 che ha permesso ai prodotti già presenti sul mercato alla data del 6 giugno 1995 di rimanere in commercio, in virtù di una disposizione ope legis. Tali disposizioni transitorie sono terminate il 31 dicembre 2019.
 La legge n.190/2014 ha disposto che, per ottenere un rinnovo dell’autorizzazione alla commercializzazione, il titolare dovesse depositare in AIFA i dossier di registrazione entro il 30 giugno 2017. L’Agenzia, in caso di valutazione favorevole del dossier, concede un’autorizzazione all’immissione in commercio.
La legge n.190/2014 ha disposto che, per ottenere un rinnovo dell’autorizzazione alla commercializzazione, il titolare dovesse depositare in AIFA i dossier di registrazione entro il 30 giugno 2017. L’Agenzia, in caso di valutazione favorevole del dossier, concede un’autorizzazione all’immissione in commercio.
Con Legge 160/2019 è stato stabilito il proseguo della commercializzazione per i prodotti interessati da un procedimento di richiesta di AIC depositata all’ AIFA entro il 30 giugno 2017, mentre per gli altri prodotti, per i quali il titolare non ha presentato la domanda di rinnovo, è stato concesso lo smaltimento delle scorte presenti nel canale distributivo alla data del 1 gennaio 2020, fino alla data di scadenza indicata in etichetta e comunque non oltre il 1 gennaio 2022.
Aggiornamento elenchi medicinali omeopatici
Nota:
D.Lgs. 219/06 – Capo II
NORME SPECIALI APPLICABILI AI MEDICINALI OMEOPATICI
Art. 16.
Procedura semplificata di registrazione
1. Un medicinale omeopatico e’ soggetto, ai fini dell’immissione in commercio, ad una procedura semplificata di registrazione, soltanto se il medicinale:
a) e’ destinato ad essere somministrato per via orale od esterna;
b) non reca specifiche indicazioni terapeutiche sull’etichetta o tra le informazioni di qualunque tipo che si riferiscono al prodotto;
c) ha un grado di diluizione tale da garantirne la sicurezza; in ogni caso il medicinale non può contenere più di una parte per diecimila di tintura madre, ne’ più di 1/100 della più piccola dose eventualmente utilizzata nell’allopatia per le sostanze attive la cui presenza in un medicinale allopatico comporta l’obbligo di presentare una ricetta medica.
2. Con decreto del Ministro della salute sono adottati eventuali nuovi parametri concernenti la sicurezza del medicinale omeopatico in sostituzione o a integrazione di quelli previsti dalla lettera c) del comma 1, conformemente a quanto stabilito dalla Comunità europea.
3. Al momento della registrazione, l’AIFA stabilisce il regime di fornitura del medicinale.
4. Le disposizione degli articoli 8, comma 3, 29, comma 1, da 33 a 40, 52, comma 8, lettere a), b) e c), e 141 si applicano, per analogia, alla procedura semplificata di registrazione dei medicinali omeopatici, ad eccezione delle prove di efficacia terapeutica.
Art. 17.
Contenuto della domanda di registrazione semplificata
1. La domanda di registrazione semplificata può riguardare una serie di medicinali ottenuti dagli stessi materiali di partenza per preparazioni omeopatiche o ceppi omeopatici.
2. In ogni caso la domanda di registrazione semplificata, da presentare conformemente ad uno specifico modello stabilito dall’AIFA entro tre mesi dalla data di entrata in vigore del presente decreto e pubblicato nella Gazzetta Ufficiale della Repubblica italiana, deve contenere ed essere corredata dei seguenti dati e documenti diretti, in particolare, a dimostrare la qualità farmaceutica e l’omogeneità dei lotti di produzione:
a) denominazione scientifica del materiale o dei materiali di partenza per preparazioni omeopatiche o ceppi omeopatici o altra denominazione figurante in una farmacopea, con l’indicazione delle diverse vie di somministrazione, forme farmaceutiche e gradi di diluizione da registrare;
b) denominazione propria della tradizione omeopatica;
c) dossier che descrive le modalità con cui si ottiene e si controlla ciascun materiale di partenza per preparazioni omeopatiche o ceppo omeopatico e ne dimostra l’uso omeopatico mediante un’adeguata bibliografia;
d) documentazione concernente i metodi di produzione e di controllo per ogni forma farmaceutica e una descrizione dei metodi di diluizione e dinamizzazione;
e) autorizzazione alla produzione dei medicinali oggetto della domanda;
f) copia di ogni registrazione o autorizzazione eventualmente ottenuta per lo stesso medicinale in altri Stati membri della Comunità europea;
g) un modello dell’imballaggio esterno e del confezionamento primario dei medicinali da registrare;
h) dati concernenti la stabilità del medicinale.
Art. 18.
Medicinali omeopatici a cui non si applica la procedura semplificata di registrazione
1. I medicinali omeopatici diversi da quelli a cui si riferisce l’articolo 16, comma 1, devono essere autorizzati ed etichettati conformemente agli articoli 8, 10, 11, 12, 13 e 14. Nei riguardi della documentazione presentata a sostegno della domanda si applica il disposto dell’articolo 8, comma 4. Per tali prodotti possono essere previste, con decreto del Ministro della salute, su proposta dell’AIFA, norme specifiche relative alle prove precliniche e alle sperimentazioni cliniche, in coerenza con i principi e le caratteristiche della medicina omeopatica praticata in Italia.
2. Il titolo IX del presente decreto si applica ai medicinali omeopatici, ad eccezione di quelli ai quali si riferisce l’articolo 16, comma 1.
Art. 19.
Comunicazioni in ambito comunitario
1. L’AIFA comunica agli altri Stati membri della Comunità europea ogni informazione necessaria a garantire la qualità e l’innocuità dei medicinali omeopatici prodotti e immessi in commercio nella Comunità europea.
Art. 20.
Disposizione transitoria sui medicinali omeopatici; estensione della disciplina ai medicinali antroposofici
1. Per i medicinali omeopatici presenti sul mercato italiano alla data del 6 giugno 1995, resta fermo quanto previsto dalla normativa vigente alla data di entrata in vigore del presente decreto; l’autorizzazione o la registrazione semplificata di tali prodotti e’ rilasciata secondo la disciplina del presente decreto.
2. Ai prodotti di cui al comma 1 si applicano, in ogni caso, le disposizioni previste dal titolo IX [ndr: farmacovigilanza] .
3. I medicinali antroposofici descritti in una farmacopea ufficiale e preparati secondo un metodo omeopatico sono assimilabili, agli effetti del presente decreto, ai medicinali omeopatici.




