L’Area Autorizzazioni Medicinali (AAM) dell’Agenzia Italiana del Farmaco (AIFA) gestisce i procedimenti relativi alle seguenti attività:
 Autorizzazioni all’Immissione in Commercio (AIC) di medicinali per uso umano contenenti sostanze attive di sintesi chimica e di origine biologica, presentate con procedura nazionale o di mutuo riconoscimento e decentrata;
Autorizzazioni all’Immissione in Commercio (AIC) di medicinali per uso umano contenenti sostanze attive di sintesi chimica e di origine biologica, presentate con procedura nazionale o di mutuo riconoscimento e decentrata;- modifiche, trasferimenti di titolarità e rinnovi delle AIC;
- importazione parallela dei medicinali;
- procedure di importazione ed esportazione del sangue umano e dei suoi prodotti destinati alla produzione di medicinali emoderivati;
- certificazione di Plasma Master File (PMF) nazionale.
Le competenze dell’Area Autorizzazioni Medicinali e degli Uffici ad essa afferenti sono definite nel Regolamento di organizzazione dell’AIFA: https://www.aifa.gov.it/documents/20142/629739/Regolamento_AIFA_2016_3.pdf.
Per essere commercializzati in Italia, i medicinali devono ricevere un’AIC da parte dell’AIFA. A tale scopo le aziende farmaceutiche sono tenute a presentare una domanda di autorizzazione, corredata da documentazione idonea e conforme alla normativa nazionale ed europea in materia di medicinali e agli standard di qualità internazionali.
La domanda deve includere informazioni su:
- sviluppo del medicinale e sua caratterizzazione;
- processo di fabbricazione;
- requisiti per il controllo della qualità e della stabilità della sostanza attiva e del prodotto finito.
Se la sostanza attiva è già descritta nella Farmacopea Europea, in sostituzione della documentazione pertinente alle relative sezioni del dossier può essere presentato, ove disponibile, un certificato di conformità alla monografia di Farmacopea (Certificate of Suitability – CEP), rilasciato dalla Direzione Europea della Qualità dei Medicinali (EDQM).
In alternativa, il richiedente l’AIC può far riferimento a un Active Substance Master File (ASMF), un documento separato dal dossier autorizzativo, che il produttore del principio attivo deposita presso l’AIFA e che contiene informazioni dettagliate, confidenziali, sul processo produttivo e sul controllo di qualità della sostanza attiva.
A supporto della domanda di AIC devono essere presentati, inoltre, dati che dimostrino l’efficacia e la sicurezza del medicinale o l’equivalenza, in termini di efficacia e sicurezza, rispetto a un altro già autorizzato.
I dati da presentare possono variare secondo la base giuridica della domanda, ovvero in relazione al riferimento normativo scelto per la domanda di autorizzazione, in accordo al Decreto Legislativo 24 aprile 2006, n. 219 (Attuazione della direttiva 2001/83/CE -e successive direttive di modifica- relativa ad un codice comunitario concernente i medicinali per uso umano, nonché’ della direttiva 2003/94/CE, artt. 8.3, 10.1, 10.6, 11, 12, 13, 21) e al Regolamento (CE) n. 726/2004 del Parlamento europeo e del Consiglio del 31 marzo 2004, che istituisce procedure comunitarie per l’autorizzazione e la sorveglianza dei medicinali per uso umano e veterinario e che segna l’istituzione dell’Agenzia Europea per i Medicinali (art. 6).
Il richiedente deve inoltre presentare la strategia di gestione del rischio in relazione ai potenziali effetti indesiderati, non noti o attesi per il medicinale, sulla base degli studi clinici condotti in fase di registrazione, e proporre le informazioni del prodotto da fornire ai medici e ai pazienti per l’utilizzo corretto del farmaco (Riassunto delle Caratteristiche del Prodotto – RCP, Foglio Illustrativo – FI, materiale di confezionamento).
Le procedure per presentare una domanda di AIC si distinguono in:
- nazionali;
• di mutuo riconoscimento (MR); - • decentrate (DC);
• centralizzate.
È il richiedente a scegliere quale procedura intraprendere, fatta salva l’obbligatorietà di seguire quella centralizzata, gestita dall’Ufficio Procedure Centralizzate dell’AIFA, per alcune categorie di medicinali identificate dalla normativa europea. L’Ufficio AIC gestisce le domande di AIC presentate con proce- dura nazionale, di Mutuo Riconoscimento (MR) e Decentrata (DC).
Per le procedure nazionali l’AIFA rappresenta l’unica Autorità competente responsabile del procedi- mento e può rilasciare un’autorizzazione valida esclusivamente nel territorio italiano.
Per le altre procedure europee la valutazione è eseguita in collaborazione con le Autorità Competenti dell’Unione Europea e l’Italia può agire come Stato Membro di Riferimento (Reference Member State- RMS), che redige il rapporto di valutazione, o Stato Membro Interessato (Concerned Member State- CMS), che verifica e commenta il rapporto redatto dal RMS. Il medicinale è quindi valutato, ed eventualmente approvato, da tutti gli Stati coinvolti nella procedura.
I dati presentati a supporto della domanda di AIC vengono valutati secondo i requisiti regolatori vi- genti e le linee guida scientifiche dell’EMA , dell’International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH)e della Commissione Europea. Il medicinale è autorizzato solo se ne vengono dimostrate la qualità, l’efficacia e la sicurezza per i soggetti per i quali l’uso è raccomandato, ovvero solo se i benefici del medicinale superano i rischi.
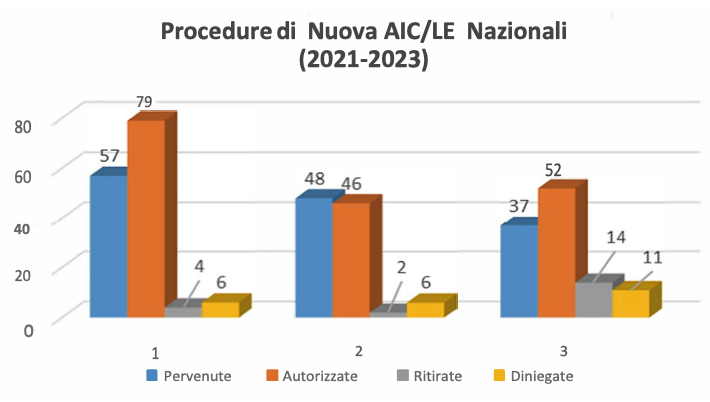
 L’iter di autorizzazione, dalla presentazione della domanda alla valutazione amministrativa e tecnica della documentazione fino all’emissione del provvedimento finale di autorizzazione e alla pubblica- zione della Relazione Pubblica di Valutazione (Public Assessment Report – PAR) sul sito dell’AIFA, segue le disposizioni previste dalla normativa, garantendo la trasparenza dell’azione amministrativa.
L’iter di autorizzazione, dalla presentazione della domanda alla valutazione amministrativa e tecnica della documentazione fino all’emissione del provvedimento finale di autorizzazione e alla pubblica- zione della Relazione Pubblica di Valutazione (Public Assessment Report – PAR) sul sito dell’AIFA, segue le disposizioni previste dalla normativa, garantendo la trasparenza dell’azione amministrativa.
A seguito dell’ottenimento dell’AIC, il titolare può presentare una richiesta di estensione di linea (line extension – LE) per l’autorizzazione di un nuovo dosaggio, di una nuova forma farmaceutica o via di somministrazione di un medicinale. Un’estensione di linea è valutata e concessa in conformità alla stessa procedura applicata per l’AIC iniziale.
Ulteriori informazioni sui requisiti regolatori delle domande di AIC dei medicinali di sintesi chimica o di origine biologica e sulle procedure di autorizzazione afferenti all’Ufficio Autorizzazione all’Immis- sione in Commercio sono disponibili sul sito dell’Agenzia ai seguenti link:
https://www.aifa.gov.it/procedura-di-autorizzazione-nazionale
https://www.aifa.gov.it/procedura-di-autorizzazione-di-mutuo-riconoscimento-e-decentrata
Tempi di autorizzazione
La norma dispone che le procedure di AIC vengano valutate in 210 giorni, esclusi i tempi interlocutori necessari alle aziende per finalizzare le richieste di integrazione e di chiarimenti emesse in corso di valutazione. Per le procedure di Mutuo Riconoscimento, relative a medicinali già autorizzati almeno in uno Stato Membro, i tempi si riducono a 60 giorni, estendibili a 90 in particolari circostanze. Dopo l’approvazione del medicinale è prevista un’ulteriore fase per l’attribuzione del regime di fornitura da parte della Commissione Tecnico Scientifica (CTS) dell’AIFA, nei casi applicabili.
Per i medicinali di eccezionale rilevanza terapeutica o utilizzabili esclusivamente in ambiente ospe- daliero la domanda di classificazione ai fini della rimborsabilità può essere presentata prima del rila- scio del provvedimento di AIC.
Per le procedure Decentrate e di Mutuo Riconoscimento è prevista, prima dell’emissione del provvedimento di autorizzazione, una fase nazionale di 30 giorni in cui, oltre alla definizione del regime di fornitura, sono valutate le traduzioni in italiano delle informazioni del prodotto.
L’AIFA pubblica sul proprio sito web il Riassunto delle Caratteristiche del Prodotto e il Foglio Illustrativo di ogni medicinale, nonché lo stato amministrativo dello stesso (se autorizzato o revocato o sospeso), nella Banca Dati Farmaci, l’unica banca dati ufficiale che permette la consultazione delle informazioni sul prodotto dei farmaci autorizzati in Italia. Per approfondimenti si rimanda ai seguenti link:
https://www.aifa.gov.it/trova-farmaco https://www.aifa.gov.it/regime-di-fornitura-dei-farmaci
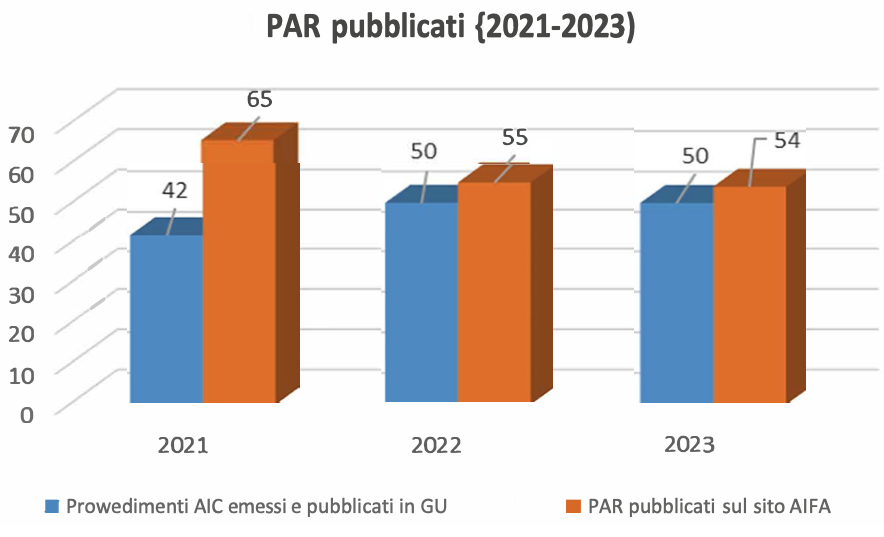
Informazioni pubbliche sulla valutazione dei medicinali
Sul sito dell’Agenzia sono pubblicate le Relazioni Pubbliche di valutazione (PAR) per i medicinali ap- provati attraverso la procedura nazionale e un riassunto in italiano dei PAR relativi ai farmaci approvati attraverso procedure di MR e DC quando l’Italia agisce in qualità di Stato Membro di Riferimento, pubblicati integralmente in inglese nel portale comune delle Agenzie Europee (Heads of Medicines, HMA), all’interno della sezione MRI product index. Si rimanda ai seguenti link:
https://www.aifa.gov.it/par-aic-nazionali
https://www.aifa.gov.it/par-italia-rms
Per tutto il ciclo di vita dei medicinali autorizzati in Italia l’AIFA assicura che la qualità e le informazioni di sicurezza approvate siano sempre aggiornate e in linea con lo stato attuale delle conoscenze scientifiche.Pertanto, successivamente alla prima autorizzazione, un’AIC può essere modificata, sospesa, 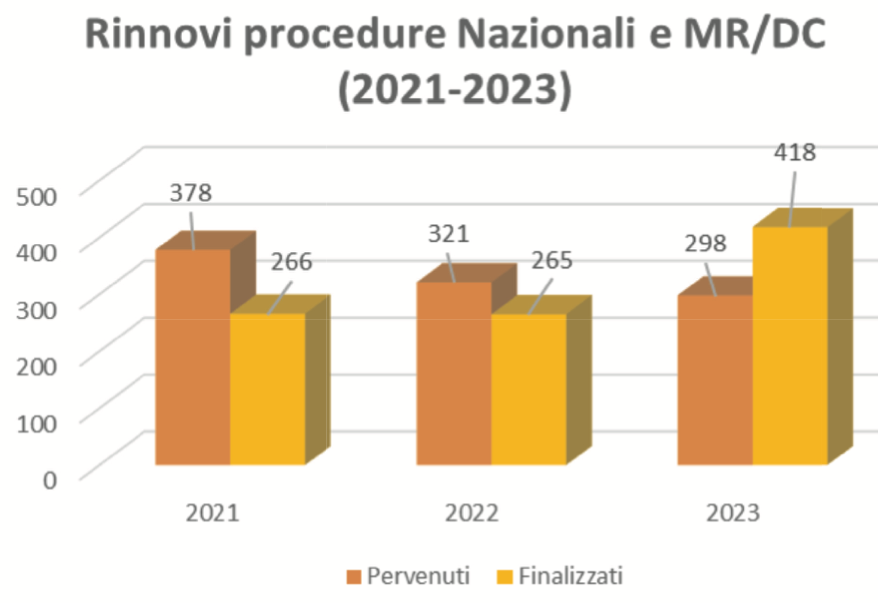 rinnovata o revocata.
rinnovata o revocata.
Dopo cinque anni dal rilascio della prima autorizzazione all’immissione in commercio, l’AIC deve essere rinnovata. A seguito del rinnovo essa ha una durata illimitata, fatta salva la possibilità di revocarla su richiesta del titolare o per motivi di sicurezza, nel caso in cui dovesse emergere da nuovi dati che il rapporto beneficio/rischio non è più favorevole.
L’Ufficio PPA pubblica sul sito dell’AIFA l’elenco dei medicinali per i quali l’AIC decade ope legis per mancata presentazione della domanda di rinnovo.
Ulteriori informazioni sui rinnovi sono disponibili al link:
https://www.aifa.gov.it/modifiche-rinnovi-e-decadenze-delle-aic
Fonte AIFA. Estratto da: Relazione annuale dell’Area Autorizzazioni Medicinali – Anno 2023
Notizie correlate: Procedura di autorizzazione centralizzata